










Un médicament biologique ou biomédicament est un médicament dont la substance active est produite ou extraite à partir d'une source biologique (cellule ou organisme vivant). Parmi ces biomédicaments on retrouve notamment : les vaccins, les facteurs de croissance, les médicaments dérivés du sang ou encore les anticorps monoclonaux.
Cancérologie, rhumatologie, gastro-entérologie, diabétologie ou encore dermatologie, nombreux sont les domaines thérapeutiques où les biomédicaments sont devenus le socle de la prise en charge des patients, bien souvent atteints de pathologies lourdes.
Certains de ces médicaments biologiques tels que les anticorps monocolonaux font partie des produits pharmaceutiques les plus utilisés et occasionnent une dépense très importante, tant à l'hôpital qu'en ville. À l'hôpital par exemple, le bevacizumab a occasionné en 2016 une dépense de l'ordre de 322,7 millions d'euros dans les établissements MCO publics et privés en France. [1]
[1] Données ScanSanté ATIH
Les médicaments biologiques similaires, plus communément appelés médicaments biosimilaires, sont des « copies » de biomédicaments de référence pouvant être commercialisées lorsque leur brevet est tombé dans le domaine public.
D'après l'article L5121-1-15 du Code de la Santé Publique, il s'agit d' « un médicament biologique de même composition qualitative et quantitative en substance active et de même forme pharmaceutique qu'un médicament biologique de référence, mais qui ne remplit pas les conditions pour être regardé comme une spécialité générique en raison de différences liées notamment à la variabilité de la matière première ou aux procédés de fabrication et nécessitant que soient produites des données précliniques et cliniques supplémentaires dans des conditions déterminées par voie réglementaire. »
Un cadre réglementaire spécifique aux médicaments biosimilaires a été établi en Europe dès 2005, au Japon en 2009, et plus récemment aux États-Unis, en 2012. Dans les pays de l'union européenne, les demandes d'autorisation de mise sur le marché (AMM) des médicaments biosimilaires sont soumises à une procédure d'examen centralisée par l'Agence Européenne du Médicament. Cette dernière a émis des recommandations spécifiques aux médicaments biosimilaires, précisant les pré-requis pour l'obtention d'une AMM. Un exercice de comparabilité incluant des essais précliniques et des essais cliniques de phase I et de phase III est nécessaire. Le rigoureux procédé d'évaluation mis en place permet de garantir que le médicament biosimilaire ne présente pas de différences significatives avec le biomédicament de référence en termes de qualité, d'efficacité et de sécurité. [1]
Fiche de bon Usage de la HAS (novembre 2017)
[1] Beck M, Michel B, Rybarczyk-Vigouret MC, Levêque D, Sordet C, Sibilia J, Velten M. Les médicaments biosimilaires : quels enjeux pour les professionnels de santé ? mt 2016 ; 22 (6) : 354-63 doi:10.1684/met.2016.0595
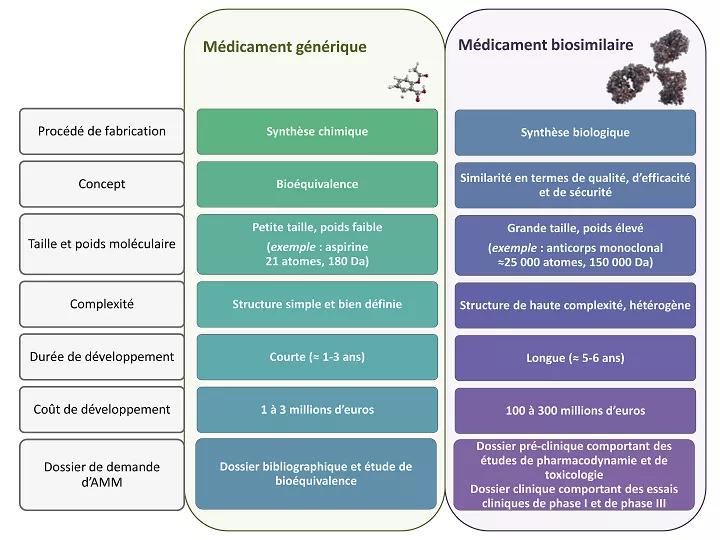
Les médicaments biosimilaires s’apparentent aux médicaments génériques sur le principe de la perte d’exclusivité des laboratoires pharmaceutiques, ouvrant le marché à la concurrence. En revanche, il s’agit là certainement de la seule analogie.
La complexité de ces produits et de leur procédé de fabrication est telle qu'ils ne peuvent être strictement identiques au biomédicament de référence.
Médicaments génériques et biosimilaires : quelles différences ?
Médicaments biosimilaires commercialisés en France avec codes UCD / CIP / CIS (Aout 2024) :
Une autre question prégnante concerne la possibilité de passer d'un traitement par un biomédicament de référence à un médicament biosimilaire, ou encore de passer d'un médicament biosimilaire à un autre biosimilaire. Il s'agit d'une notion d'interchangeabilité si l'échange relève d'un acte de prescription réalisé sous la responsabilité du médecin, et d'une notion de substitution si l'échange relève d'un acte de délivrance réalisé par un pharmacien. Contrairement à l'AMM, laquelle est obtenue à l'échelle européenne, l'EMA laisse à chaque État membre le soin de définir ses propres règles en matières d'interchangeabilité et de substitution.
En France, l'Agence nationale de sécurité du médicament et des produits de santé (ANSM) avait initialement émis la recommandation de ne pas remplacer un biomédicament par un autre en cours de traitement. L'évolution des connaissances et les données d'efficacité et de sécurité rassurantes au sein de l'UE l'ont toutefois amenée à réviser dernièrement sa position sur l'interchangeabilité des médicaments biosimilaires.:
Dans ce rapport publié en mai 2016, l'agence sanitaire française indique qu'une interchangeabilité en cours de traitement est envisageable sous trois conditions :
- L'information et l'obtention de l'accord du patient ;
- La mise en place d'une surveillance clinique appropriée ;
- L'assurance de la traçabilité des produits concernés.
- L'information et l'obtention de l'accord du patient ;
- La mise en place d'une surveillance clinique appropriée ;
- L'assurance de la traçabilité des produits concernés.
Ce positionnement intervient dans le cadre d'une réflexion sur le décret d'application de l'article 47 de la loi de financement de la Sécurité sociale pour 2014 (LFSS) lequel statue que le pharmacien est autorisé à substituer à un biomédicament de référence un médicament biosimilaire appartenant au même groupe biologique similiaire, uniquement dans le cas d'une initiation de traitement et si le prescripteur n'a pas exclu cette possibilité de substitution.
L'Omédit Grand Est, en partenariat avec l'Assurance Maladie Grand Est et l'ARS Grand Est, a créé un document d'information sur les médicaments biosimilaires à destination des patients : "Mieux comprendre mon médicament biosimilaire".
Le CHRU de Nancy, en partenariat avec l'Omédit, a produit des 3 documents d'informations dans le cadre de son engagement à prescrire des médicaments biosimilaires :
Publications :
Qu'est ce qu'un médicament biosimilaire ? Similitudes et différence avec les médicaments génériques ? Les enjeux économiques et de santé publique ? Biosimilaires, interchangeabilité et concurrence ? Retrouvez les réponses dans les actes du colloque organisé en décembre 2017
-
Knowledge, behaviors and practices of community and hospital pharmacists towards biosimilar medicines: Results of a French web-based survey (pdf, 785.95 Ko) M. Beck, B. Michel, M.C. Rybarczyk-Vigouret, D Levêque, C Sordet, J Sibiliad, and M, Veltene, MAbs. Dec 2016
Les objectifs de cette étude étaient d'extraire un aperçu complet des connaissances, de l'expérience et des opinions des pharmaciens d'officines et des pharmaciens hospitaliers concernant les médicaments biosimilaires en France; et d'identifier les problèmes perçus et les solutions pour promouvoir leur prescription.
-
Rheumatologists’ Perceptions of Biosimilar Medicines Prescription: Findings from a French Web-Based Survey (pdf, 608.03 Ko) M. Beck, B. Michel, M.C. Rybarczyk-Vigouret, D. Levêque, C. Sordet, J. Sibilia, M. Velten; BioDrugs. 2016 Dec;30(6):585-592.
Le but de cette étude était d'évaluer les connaissances, l'expérience et les opinions des rhumatologues français hospitaliers et libéraux en ce qui concerne les médicaments biosimilaires et d'identifier les obstacles et les options possibles pour promouvoir leur prescription.
-
Biosimilar infliximab for the management of rheumatoid arthritis in France: what are the expected savings? (pdf, 343.53 Ko) M. Beck, B. Michel, M.C. Rybarczyk-Vigouret, C. Sordet, J. Sibilia, M. Velten ; European Journal of Hospital Pharmacy
Le biosimilaire de l’infliximab, le premier médicament biologique similaire contenant des anticorps monoclonaux à être commercialisé, devrait contribuer à une réduction significative des coûts de santé. Nous avons cherché à évaluer le potentiel d'économies des dépenses de santé avec l'utilisation du biosimilaire pour le traitement des patients atteints de polyarthrite rhumatoïde (PR) sur plus d'un an dans les conditions de vie réelles en Alsace et en France.
-
Les médicaments biosimilaires : quels enjeux pour les professionnels de santé ? (pdf, 431.04 Ko) M. Beck, B. Michel, M.C. Rybarczyk-Vigouret, D. Levêque, C. Sordet, J. Sibilia, M. Velten ; Médecine thérapeutique, 2016
Posters :